You are successfully logged out.
Trodelvy® is the first and only approved Trop-2-directed antibody-drug conjugate that selectively delivers a cytotoxic payload, SN-38, to cancer cells, including triple-negative and HR+/HER2- metastatic breast cancer cells, and the tumor microenvironment through a bystander effect.1,4

Trodelvy® works by targeting Trop-2 to deliver SN-38 to triple-negative breast cancer tumours1

The proposed MOA is based on preclinical data, which may not correlate with clinical outcomes.
Trodelvy® binds to Trop-2 which is overexpressed in a high percentage of triple-negative breast cancer tumors, but with limited expression in normal tissue.2,4,5
Trodelvy® carries SN-38 (active metabolite of irinotecan), a topoisomerase 1 inhibitor.5
Allows Trodelvy® to deliver a higher concentration of SN-38 to triple-negative breast cancer cells and microenvironment.5
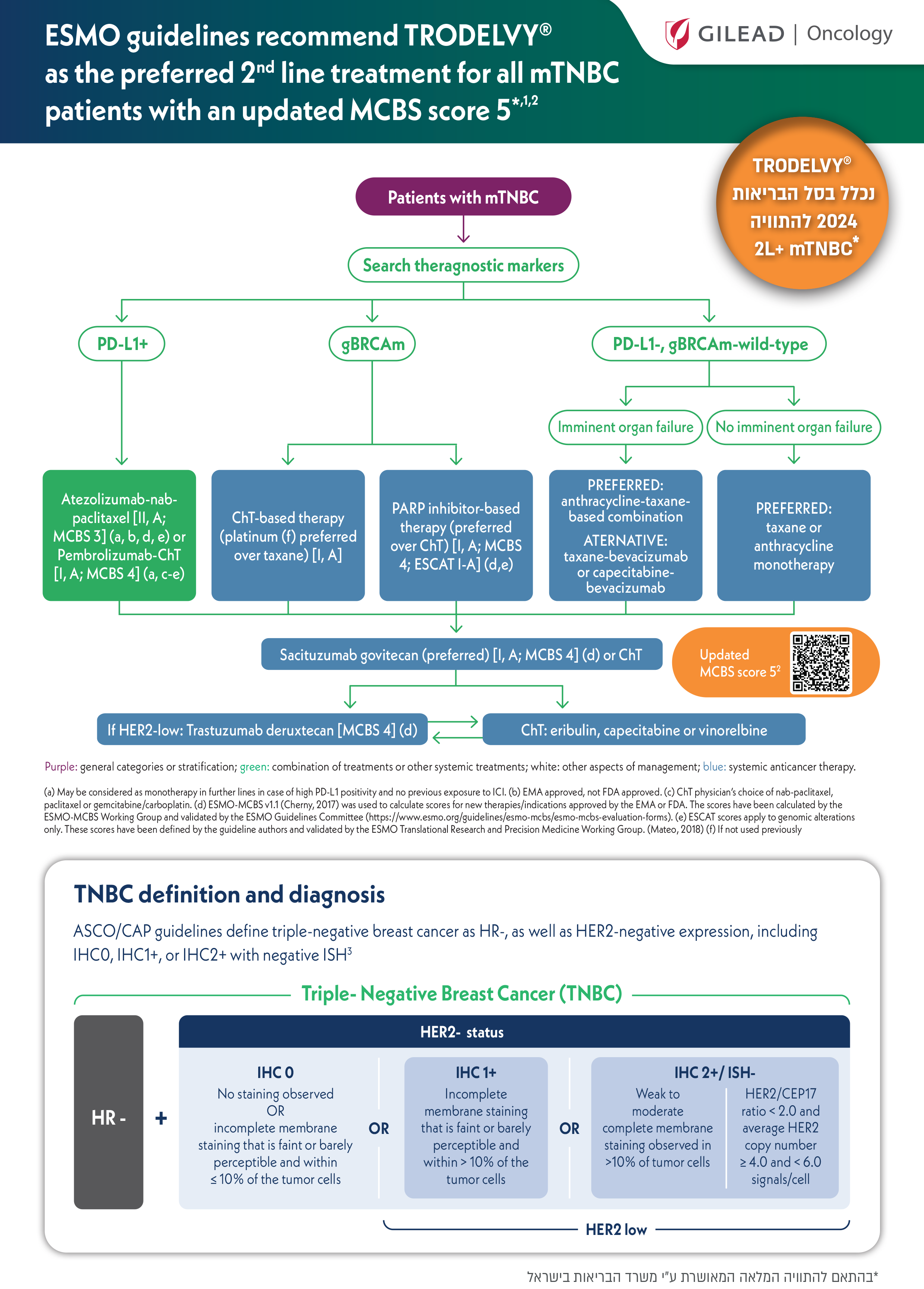
Find out where Trodelvy® fits into treatment for adult patients progressing from earlier stage or refractory/relapsed metastatic triple-negative breast cancer or diagnosed with de novo unresectable locally advanced or metastatic triple-negative breast cancer.
These pathways relate to the proposed positioning of Trodelvy® with respect to its approved indication by the Israeli Ministry of Health and are not intended as a comprehensive representation of guidelines for unresectable locally advanced or metastatic triple-negative breast cancer.
Proposed treatment pathway for adult patients progressing from earlier stage or refractory/relapsed metastatic triple-negative breast cancer.*1

*Earlier adjuvant or neoadjuvant treatment for more limited disease will qualify as one of the required prior regimens.1
Proposed treatment pathway for adult patients diagnosed with De novo metastatic triple-negative breast cancer.1

Trodelvy® significantly improved survival in as early as second-line or later metastatic triple-negative breast cancer when compared to single-agent chemotherapy.2*
The ASCENT trial was an international, multicentre, open-label, randomised phase 3 study to investigate Trodelvy® in patients with unresectable locally advanced or metastatic triple-negative breast cancer who had relapsed after at least 2 prior lines of chemotherapy (one of which could be in the neoadjuvant or adjuvant setting provided progression occurred within a 12-month period after completion of treatment).2

*The proportion of patients with known brain metastases at baseline was capped at 15%.2
**PFS was assessed by blinded independent central review based on RECIST 1.1 criteria in patients without known brain metastases (n=468).2
In ASCENT, demographics and baseline characteristic were well matched between groups2

Median progression free survival (PFS) with Trodelvy® was nearly three times longer than with single-agent
chemotherapy (ITT population)*

*Assessed by independent central review in the ITT population. The improvements in PFS in the primary analysis population were consistent with the ITT population (median PFS: 5.6 months vs 1.7 months; HR: 0.41; P<.0001, with TRODELVY® vs Single-agent chemotherapy respectively).
Median overall survival (OS) with Trodelvy® was nearly 1 year (ITT population)*

*Assessed by independent central review in the ITT population. The improvements in OS in the primary analysis population were consistent with the ITT population (median OS: 12.1 months vs 6.7 months; P<.0001 with TRODELVY® vs Single-agent chemotherapy respectively).
Trodelvy® delivered more than seven times greater objective response rate than single-agent
chemotherapy2 (ITT population)*

4% with TRODELVY vs 1% with single-agent chemotherapy
27% with TRODELVY vs 3% with single-agent chemotherapy
40% with TRODELVY vs 8% with single-agent chemotherapy
6.3 months with TRODELVY vs 3.6 months with single-agent chemotherapy
*Based on ORR for TRODELVY and single-agent chemotherapy as shown in Table 3 of the Summary of Product Characteristics.1
†Assessed by independent central review in the ITT population. The ORR results in the primary analysis population (ORR: 35% vs 5%; OR: 10.8; 95% CI, 5.6–21.0) were consistent with the ITT population.2 The primary analysis population consisted of patients without present or prior history of brain metastases (n=468). The ITT population consisted of patients with or without brain metastases at baseline (N=529).1
‡Assessed by independent central review in the ITT population.2 CR defined as disappearance of all target lesions. Any pathological lymph nodes (whether target or non-target) must have reduction in short axis to <10 mm.3
§Assessed by independent central review in the ITT population.2 PR defined as at least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters.3
∥Assessed by independent central review in the ITT population. Clinical benefit rate defined as a CR, a PR, or stable disease with a duration of at least 6 months.2
CI, confidence interval; CR, complete response; ITT, intent-to-treat; OR,
odds ratio; ORR, objective response rate; PR, partial response.
*Assessed by independent central review in the ITT population. The ORR results in the primary analysis population were consistent with the ITT population (ORR: 35% vs 5%; OR: 10.8; 95% CI, 5.6–21.0 with TRODELVY® vs Single-agent chemotherapy respectively).
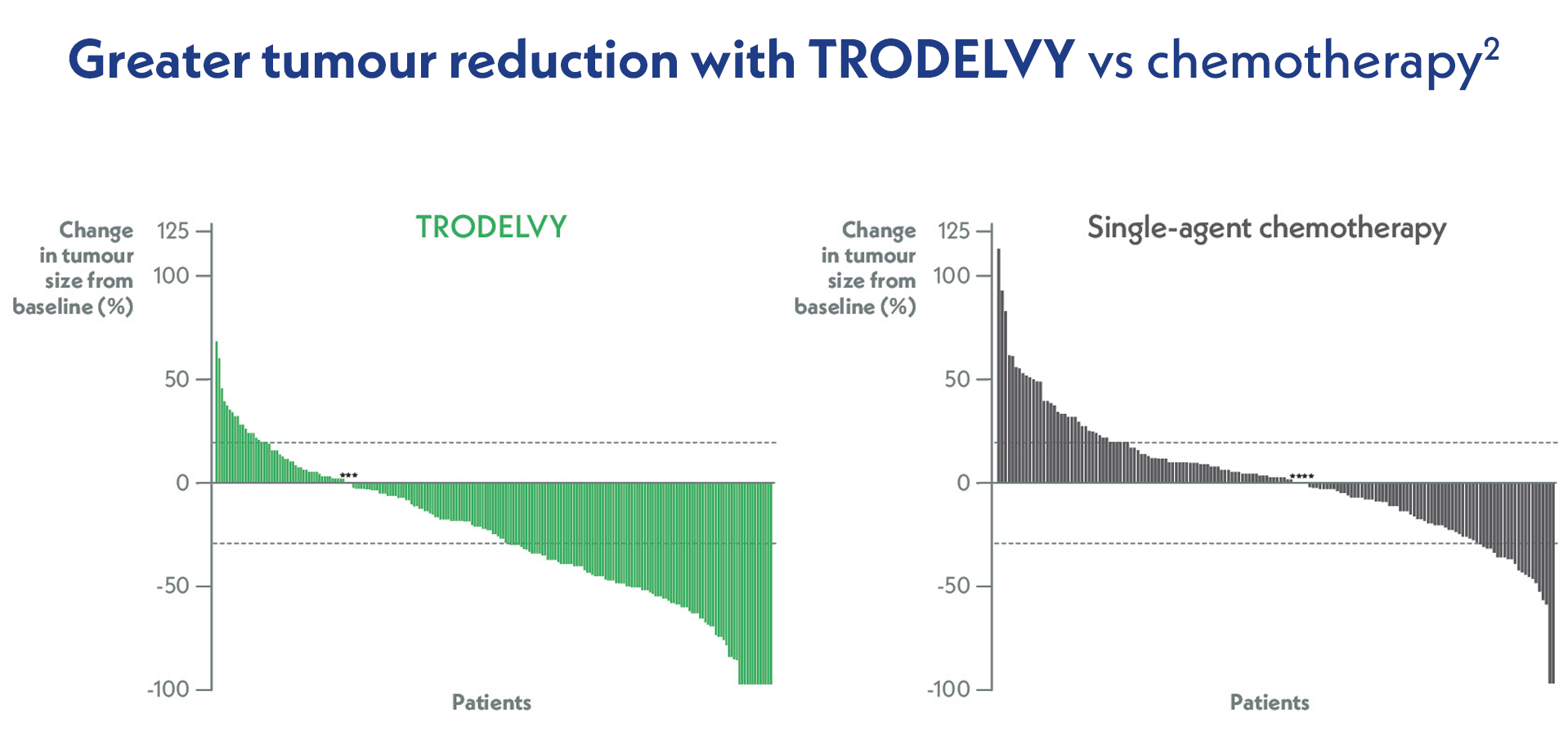
Adapted from Bardia A, et al. N Engl J Med. 2021.
Waterfall plot of the best percent change in the sum of the diameters of target lesions in patients without brain metastases who had at least one response assessment by central review (n=212 patients in the TRODELVY group and n=160 in the chemotherapy group). Asterisks at 0 denote patients who had no change from baseline in tumour size.2
In an assessment of patient-reported outcomes in the ASCENT trial, Trodelvy was generally associated with greater improvements and delayed worsening of HRQoL scores compared with TPC.6
Trodelvy demonstrated superiority versus TPC in the following primary HRQoL domains6:
TPC-treatment of physician choice
In the phase 3 ASCENT trial of patients with metastatic triple-negative breast cancer:
Adverse events of special interest in the ASCENT trial2

This is not an exhaustive list. For full details of adverse events, please refer to the Trodelvy® prescription information approved by Israeli Ministry of Health.1
*Combined preferred terms of “Neutropenia” and “Decreased neutrophil count”.2 **Combined preferred terms of “Anaemia”, “Decreased haemoglobin” and “Decreased red-cell count”.2 †Combined preferred terms of “Leukopenia” and “Decreased white blood cell count”.2
The most common adverse reactions reported in patients treated with sacituzumab govitecan were: neutropenia (67.6%), nausea (62.6%), diarrhoea (62.5%), fatigue (61.5%), alopecia (45.6%), anaemia (40.7%), constipation (36.2%), vomiting (33.6%), decreased appetite (25.7%), dyspnoea (22.1%) and abdominal pain (20.2%).1
Adverse events leading to discontinuation were infrequent in both arms of the study

*Combined preferred terms of “neutropenia” and “decreased neutrophil count”.2
Learn how to appropriately dose and administer Trodelvy®*.


The infusion rate of TRODELVY should be slowed down or infusion interrupted if the patient develops an infusion-related reaction. Grade ≥3 infusion reactions occurred in 1.9% of patients receiving TRODELVY (n=7/366).
Consider antiemetic preventive treatment with 2 or 3 medicinal products to prevent and treat chemotherapy-induced nausea and vomiting (CINV), e.g.:
Pre-infusion medication is recommended to prevent infusion reactions, e.g.:
*Please refer to the Approved prescribing information for a full summary of the safety profile for Trodelvy®, premedication, recommended dose modifications, and management strategies.
Adverse events in patients receiving Trodelvy® can usually be managed with dose modifications and routine management strategies.
*Please refer to the Approved prescribing information for a full summary of the safety profile for Trodelvy®, premedication, recommended dose modifications, and management strategies.
Dose modifications can be made as needed to help manage diarrhoea and other non-neutropenic toxcity1

The sacituzumab govitecan dose should not be re-escalated after a dose reduction for adverse reactions has been made
Dose modifications can be made as needed to help manage neutropenia1
Grade 4 neutropenia ≥ 7 days or less if clinically indicated,
or
Grade 3-4 febrile neutropenia
or
At time of scheduled treatment, Grade 3-4 neutropenia which delays dosing by 2
or 3 weeks for recovery to ≤ Grade 1

Administer reactive G-CSF
as soon
as clinically indicated

25%
Dose reduction (7.5 mg/kg)
administer
G-CSF as soon as clinically indicated

50%
Dose reduction (5 mg/kg)
administer
G-CSF as soon as clinically indicated

Discontinue
treatment
administer
G-CSF as soon as clinically indicated
At time of scheduled treatment, Grade 3-4 neutropenia which delays dosing Beyond 3 weeks for recovery to ≤ Grade 1
Administer reactive G-CSF
The sacituzumab govitecan dose should not be re-escalated after a dose reduction for adverse reactions has been made